El “cazador de variantes” que halló con solo 6 secuencias la que ahora domina en España
“No tenemos datos de un cambio dramático en la severidad del virus”, dice a FARO el italiano Federico Gueli, aficionado a la ciencia
variantes covid

Vigilar la evolución del coronavirus SARS-CoV-2 es como supervisar el universo. De igual forma que astrónomos amateur escudriñan el firmamento con sus telescopios y a veces descubren asteroides potencialmente peligrosos, algunos aficionados a la ciencia están ayudando a monitorizar los cambios en el virus del COVID-19. Y conviene que haya muchos ojos sobre ellos: hay más 13 millones de genomas de este coronavirus, la mayor colección de cualquier organismo en la historia de la ciencia. Hace 6 meses eran solo 10 millones. Y los sublinajes circulantes son más de medio millar, según la OMS. Este periódico ha contactado con uno de estos “cazadores de variantes”, el italiano Federico Gueli, descubridor del linaje que actualmente domina en España al 50,8% –BQ.1 y sus derivados, incluyendo BQ.1.1– y pronto será dominante a escala global.

“Encontré BQ.1 cuando tenía 6 secuencias”, cuenta a FARO Gueli por vía telefónica. “La descubrí cuando estaba en vacaciones en un sitio turístico llamado Chiesa in Valmalenco, en Lombardía”. Este amable italiano reconoce que su formación académica en esta materia “es cero”. De hecho, es adiestrador canino. “El 19% de los hallazgos astronómicos vienen de ciudadanos. Lo mismo hacemos los buscadores de variantes”, ejemplifica. “No tenemos datos actualmente de un cambio dramático en la severidad del virus”, tranquiliza Gueli, que explica que “el fenotipo del virus es todavía atenuado”.
“El 19 por ciento de los hallazgos astronómicos vienen de ciudadanos. Lo mismo hacemos los buscadores de variantes”
El italiano relata que todo empezó de la mano de una sola persona, Áine O’Toole, joven investigadora irlandesa que trabaja en Edimburgo (Escocia). Ella y Andrew Rambaut, uno de los bioinformáticos punteros del mundo, y el también británico Oliver G Pybus, biólogo, escribieron un trabajo en la revista “Nature” y crearon la nomenclatura actual de las variantes, llamada “Pango” en referencia al pangolín, posible transmisor del SARS-CoV-2 a los humanos. Al principio era O’Toole quien asignaba los nombres manualmente mirando a los árboles, un trabajo inmenso. Y entonces llegó la “era de las variantes de preocupación”.

El descubridor del linaje BQ.1, Federico Gueli, con su primer “post” sobre el mismo en la página web de Pango. La foto está tomada en Chiesa Valmalenco, en los Alpes italianos, donde descubrió este sublinaje, que pronto será dominante en el mundo.
Era diciembre de 2020 cuando la vigilancia genómica de rutina de Reino Unido detectó lo que fue designado como linaje B.1.1.7, un grupo filogenéticamente nuevo y diferenciado. Era la variante alfa, la primera “variante de preocupación”, que causó la ola invernal de principios de 2021. Los primeros casos de alfa, sin embargo, datan de septiembre de 2020. Algo parecido pudo ocurrir con ómicron, detectada en noviembre de 2021. Un reciente estudio en “Science” –muy debatido estos días– concluyó que esta variante llevaba evolucionando durante meses en África sin ser detectada debido a la falta de vigilancia genómica, posiblemente ayudada por contagios en personas inmunodeprimidas por VIH.
Con la era de las variantes de preocupación, explica Gueli, “el equipo Pango tuvo una idea genial: abrieron el sistema de nomenclatura Pango, le dieron a la gente, a la comunidad científica y a la opinión pública, la posibilidad de proponer linajes al comité Pango, que es la autoridad que decide si una variante merece un nombre o no”. Pango fija la nomenclatura de letras y números: BA.1, BA.2, BA.5, XBB (donde la “X” indica recombinación), etc. Luego, es la OMS la que establece los nombres de variantes con letras griegas, como alfa, beta, delta y ómicron.
“Abrieron la página Github (repositorio de datos) de Pango a todo el mundo, científicos y ciudadanos: cualquiera puede proponer una variante que haya descubierto en los árboles o en un secuenciador de laboratorio. Todos los laboratorios del mundo pueden confrontar con la secuencia de referencia del virus de Wuhan. Es un método muy simple porque tenemos la herramienta”, explica Gueli, restando importancia a su labor ardua y desinteresada.

El italiano destaca el nombre del biólogo computacional Cornelius Roemer, que tuvo la idea de cómo excavar en miles y miles de secuencias que se descargan todos los días en la web virológica Gisaid. “Esto no podía hacerse solo por una persona –añade Gueli–. Nos enseñó cómo hacerlo a varios de nosotros, como yo, interesados en variantes”. Gente como Gianluca Codagnone, Shay Fleishon, Zack Hensel, Ryan Eisner, Hitoshi Sakaguchi... “Empezamos a trabajar juntos, a compartir nuestros hallazgos. Gente de Países Bajos, Italia, EE UU, Alemania, Japón... Todo el mundo haciendo su trabajo y compartiendo sus hallazgos y debatiéndolos. Ganamos técnica para reconocer mejor las secuencias y entender su evolución”, destaca Gueli.
Los nombres de los participantes del grupo central original de “buscadores de variantes” fue: Cornelius Roemer, Tom Peacock, Shay Fleishon quien lideró el radar de variantes israelí ILVR del Ministerio de Salud de Israel), Zach Hensel, Sakaguchi Hitoshi, Josette Shoenmaker, Gianluca Codagnone y Sam Schimdt, a los que se unió pronto Ryan House y un poco más tarde Nick Frohberg, Dave MacNally y Raj Rajnarayanan.
Cuando el equipo empezó con la variante delta “era como seguir huellas en el bosque –apunta Gueli–. No sabíamos dónde estaba el virus ni a dónde se dirigía”. En septiembre de 2021 contactaron con Thomas P. Peacock, virólogo del Imperial College de Londres. “Encontramos ómicron cuando había 4 secuencias, Tom Peacock la encontró. Las siguientes variantes las hallamos cuando había 4, 5 o 10 secuencias. Recuerdo haber visto la BA.5 [dominante en España y buena parte del mundo hasta hace pocos días] cuando era solo 16 secuencias. Luego encontré BQ.1 cuando tenía 6 secuencias”, asegura Gueli.
“Hacemos esto sin ninguna remuneración ni premio, y muy lejos de las organizaciones”
“Estábamos solos –añade–. El interés por el SARS-CoV-2 estaba disminuyendo y había mala actitud de la gente ante el virus y ante los científicos. Hicimos esto sin ninguna gratificación ni premio, y muy lejos de las organizaciones”, resume el italiano, que subraya la importancia de la ciencia colaborativa: “Estas herramientas las construimos ciudadanos y científicos. Pedimos mejoras a Github, lo fuimos mejorando hasta el punto de que ahora tenemos el control de la situación”, asegura.
Gueli es uno de los coautores de un trabajo sobre la evolución del SARS-CoV-2 post-ómicron publicado en Virological.org. El documento, dirigido por Thomas P. Peacock y Cornelius Roemer, advierte que otro “evento ómicron” es posible.

72 mutaciones en 2 meses
Coautor también es otro “ciudadano científico”, un profesor de instituto de Indiana (EE UU) llamado Ryan Hisner, cuyos hilos en Twitter sobre la evolución del virus han sido destacados por científicos de la talla de Eric Topol, fundador y director del instituto Scripps de California. Hisner, que descubrió la subvariante XBB.1.4.1, ha alertado sobre la aparición de secuencias que han acumulado 72 mutaciones en 2 meses, algo que atribuye a la posible acción incompleta del antiviral molnupiravir. Gueli elogia el “gran trabajo” de Hisner pero recuerda que una acumulación excesiva de mutaciones puede hacer el virus inviable. “Solo soy un cazador de variantes –apunta–. Es un problema de probabilidades, una ruleta rusa: no sabemos cómo se va a comportar el virus mutado, si va a ser capaz de transmitirse”.
Respecto a la situación actual del virus, Federico Gueli señala que BQ.1 “ha alcanzado el 40% de las secuencias globales y será dominante en todo el mundo en una o dos semanas”. Una tendencia al alza que detecta en las últimas semanas es la frecuencia de la mutación F486P en la espícula del virus, una mutación de doble nucleótido presente en linajes recombinantes como XBF y XBB.1.5, y en los “deltacron” XBC y XAY.

Los esfuerzos de secuenciación han disminuido mucho a lo largo de este año. Federico Gueli insiste en que “si el mundo quiere controlar el SARS-CoV-2 y ver que mantiene su gravedad, tiene que vigilar”.
La evolución del SARS-CoV-2 puede deparar aún muchas sorpresas, aunque una cosa es lo que sugiera la bioinformática y otra diferente las consecuencias clínicas. Como dice María de Toro, química y bioinformática española, experta en secuenciación del virus, “podemos seguir monitorizando esto hasta el infinito y más allá, pero lo que nos tiene que importar es cómo va a afectarnos con la inmunización que tenemos”. Como dice el divulgador científico Eric Topol, “todas las nuevas variantes deben considerarse inocentes hasta probarse lo contrario”.
Captura Twitter
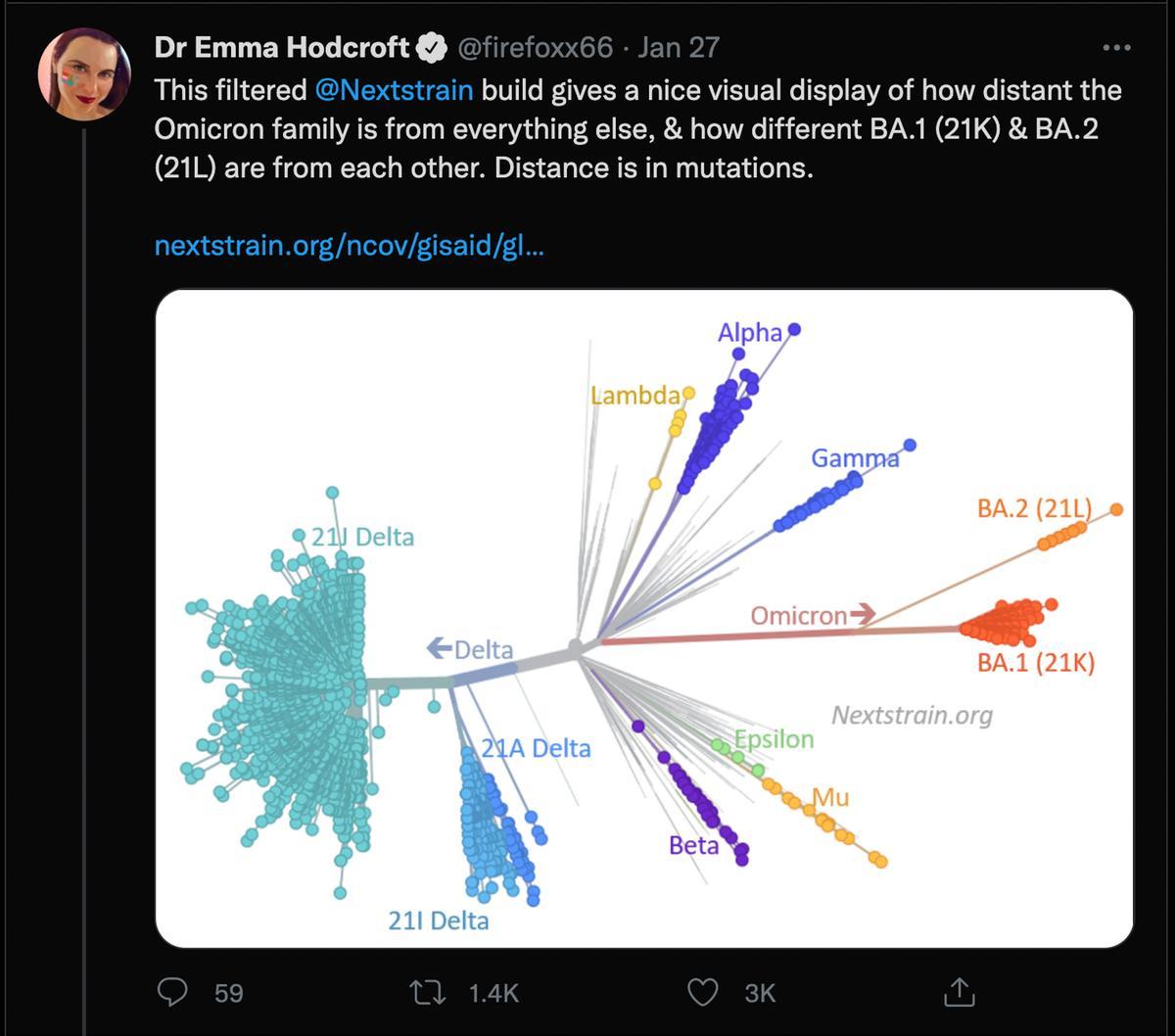
Es la primera gran pandemia con internet, y herramientas en la red como Nextstrain, Gisaid, COVID Spectrum y Taxonium permiten seguir en tiempo real la evolución del cada vez más frondoso árbol filogenético del SARS-CoV-2. Twitter permite seguir la comunicación de los hallazgos y su debate entre científicos punteros, como en este caso la epidemióloga molecular británica-estadounidense Emma Hodcroft, basada en Berna. En el tuit de la imagen muestra lo distante que es ómicron en mutaciones respecto a las variantes anteriores.
Suscríbete para seguir leyendo
- Máxima alerta de la Policía Nacional por el mensaje que está llegando a los móviles de miles de personas
- Una belleza de armas tomar
- Un nutricionista desvela qué es lo primero que le quita a los pacientes que quieren adelgazar
- Uno de los dos tripulantes gallegos desaparecidos sobrevivió al secuestro del atunero Alakrana del 2009
- El abandono no entiende de razas ni de dinero: mascotas que valen miles de euros llegan a A Madroa
- Condenan a dos años y medio de prisión al maquinista del Alvia y al exdirector de Seguridad de ADIF por el accidente de Angrois
- La familia del vigués que murió tras el concierto de Karol G anuncia denuncia: 'No hubo forcejeo ni pelea
- Mujer de más de 45 y sola busca ayuda